- For Print
- November 30, 2022
TOKYO and CAMBRIDGE, Eisai Co., Ltd. (Headquarters: Tokyo, CEO: Haruo Naito, "Eisai") and Biogen Inc. (Nasdaq: BIIB, Corporate headquarters: Cambridge, Massachusetts, CEO: Christopher A. Viehbacher, "Biogen") announced today that the results from Eisai’s large global Phase 3 confirmatory Clarity AD clinical study of lecanemab (development code: BAN2401), an investigational anti-amyloid beta (Aβ) protofibril antibody for the treatment of mild cognitive impairment (MCI) due to Alzheimer’s disease (AD) and mild AD (collectively known as early AD) with confirmed presence of amyloid pathology in the brain, were presented at the 2022 Clinical Trials on Alzheimer’s Disease (CTAD) conference, in San Francisco, California and virtually.
Summary of Presentations in the Scientific Session featuring Lecanemab at CTAD
Design of Clarity AD Study
Eisai’s Clarity AD was a global confirmatory Phase 3 placebo-controlled, double-blind, parallel-group, randomized study in 1,795 people with early AD (lecanemab group: 898 placebo group: 897) at 235 sites in North America, Europe, and Asia. The participants were randomized 1:1 to receive either placebo or lecanemab 10-mg/kg IV biweekly, and the randomization was stratified according to clinical subgroup (MCI due to AD or mild AD), presence or absence of concomitant approved AD symptomatic medication at baseline (e.g., acetylcholinesterase inhibitors, memantine, or both), ApoE4 status and geographical region. Eligibility criteria allowed patients with a broad range of comorbidities/comedications, including but not limited to hypertension, diabetes, heart disease, obesity, renal disease and anti-coagulants. As a result of Eisai’s recruitment strategy of diversity in the Clarity AD study, 4.5% and 22.5% of the randomized participants in the U.S. were Black and Hispanic, respectively.
The primary endpoint was change from baseline at 18 months in the CDR-SB1 (Clinical Dementia Rating Sum of Boxes), the global cognitive and functional scale, and key secondary endpoints were the change from baseline at 18 months in amyloid Positron Emission Tomography (PET) using Centiloids, AD Assessment Scale - Cognitive Subscale 14 (ADAS-Cog142), AD Composite Score (ADCOMS3) and AD Cooperative Study-Activities of Daily Living Scale for Mild Cognitive Impairment (ADCS MCI-ADL4). In addition, longitudinal changes in brain tau pathology as measured by tau PET (n=257) and cerebrospinal fluid (CSF) biomarkers of AD pathology (n=281) were evaluated in optional sub-studies.
Efficacy Results of Clarity AD
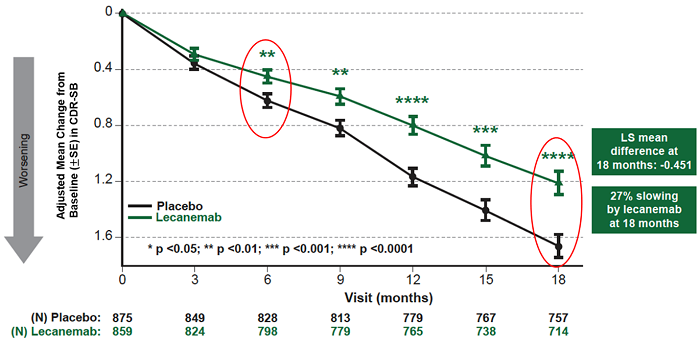
Mean change of CDR-SB from baseline at 18 months as the primary endpoint was 1.21 and 1.66 for lecanemab and placebo groups, respectively. Lecanemab treatment resulted in highly statistically significant results, reducing clinical decline on the global cognitive and functional scale, compared with placebo at 18 months by -0.45 (95% Confidence Interval (CI): -0.67, -0.23; P=0.00005), representing a 27% slowing of decline. Starting as early as six months (difference: -0.17 [95% CI: -0.29, -0.05]; P<0.01), and increasing in absolute difference over time across all time points every 3 months, the treatment showed highly statistically significant changes in CDR-SB from baseline compared to placebo (all p-values are less than 0.01) (Figure 1).
All key secondary endpoints also showed highly statistically significant results compared with placebo (P<0.001). In the amyloid PET sub-study, treatment with lecanemab showed statistically significant reduction in amyloid plaque burden at all timepoints starting at 3 months. Mean change in Centiloids at 18 months was -55.5 and 3.6 for lecanemab and placebo groups, respectively (mean difference: -59.1 [95%CI: -62.6, -55.6]; P<0.00001). Lecanemab slowed decline of cognitive function by 26% on ADAS-Cog14 at 18 months (mean difference: -1.44 [95%CI: -2.27, -0.61]; P=0.00065). In the ADCOMS assessment, lecanemab slowed disease progression by 24% at 18 months (mean difference: -0.050 [95% CI: -0.074, -0.027; P=0.00002]). Lecanemab slowed decline of activities of daily living by 37% on ADCS MCI-ADL at 18 months (mean difference: 2.016 [95%CI: 1.208, 2.823]; P<0.00001). In addition, the primary stratified analysis showed consistent results in CDR-SB, ADAS-Cog14 and ADCS MCI-ADL at 18 months of treatment with lecanemab in all subgroups of disease stage (MCI due to AD or mild AD), ApoE4 status (non-carriers, carriers), presence or absence of concomitant approved AD symptomatic medication, and region (North America, Asia, Europe).
Safety Results of Clarity AD
The most common adverse events (>10%) in the lecanemab group were infusion reactions (lecanemab: 26.4%; placebo: 7.4%), ARIA-H (combined cerebral microhemorrhages, cerebral macrohemorrhages, and superficial siderosis; lecanemab: 17.3%; placebo: 9.0%), ARIA-E (edema/effusion; lecanemab: 12.6%; placebo: 1.7%), headache (lecanemab: 11.1%; placebo: 8.1%), and fall (lecanemab: 10.4%; placebo: 9.6%). Infusion reactions were largely mild-to-moderate (grade 1-2: 96%) and occurred on the first dose (75%).
During the study period, deaths occurred in 0.7% and 0.8% of participants in the lecanemab and placebo groups, respectively and no deaths were related to lecanemab or occurred with amyloid-related imaging abnormalities (ARIA) in 18-month double-blind study period. Serious adverse events were experienced by 14.0% of participants in the lecanemab group and 11.3% of participants in the placebo group. Treatment-emergent adverse events occurred in 88.9% and 81.9% of participants in the lecanemab and placebo groups, respectively. Treatment-emergent adverse events leading to drug withdrawal occurred in 6.9% and 2.9% of participants in the lecanemab and placebo groups, respectively.
Overall, lecanemab’s ARIA incidence profile was within expectations based on the Phase 2 trial results. ARIA-E events were largely mild-to-moderate radiographically (91% of those who had ARIA-E), asymptomatic (78% of those who had ARIA-E), occurred within the first 3 months of treatment (71% of those who had ARIA-E) and resolved within 4 months of detection (81% of those who had ARIA-E). Among the 2.8% of lecanemab-treated subjects with symptomatic ARIA-E, the most commonly reported symptoms were headache, visual disturbance, and confusion. The incidence of symptomatic ARIA-H was 0.7% in the lecanemab group and 0.2% in the placebo group. No imbalance was observed in isolated ARIA-H (i.e., ARIA-H in participants who did not also experience ARIA-E) between lecanemab (8.9%) and placebo (7.8%). ARIA-E and ARIA-H were less common in ApoE4 non-carriers versus carriers, with higher frequency in ApoE4 homozygous carriers vs ApoE4 heterozygous carriers. In the core study and subsequent open-label extension study, rates of deaths with concurrent cerebral macrohemorrhage were 0.1% in both the placebo group (1/897) and the lecanemab group (2/1608). The two cases on lecanemab occurred in the open-label extension study. Both cases had significant comorbidities and risk factors including anticoagulation contributing to macrohemorrhage or death. Therefore, it is Eisai’s assessment that the deaths cannot be attributed to lecanemab.
Imaging, Plasma, and CSF Biomarkers Assessments of Clarity AD
Biomarkers assessments on amyloid, tau and neurodegeneration with lecanemab administration were conducted using imaging, plasma and CSF. Amyloid biomarkers showed early and sustained amyloid reversal effects in CSF and plasma Aβ 42/40 ratio with lecanemab treatment. Mean amyloid PET was 22.99 Centiloids at 18 months of lecanemab treatment which was below threshold for amyloid positivity of 30 Centiloids. Tau biomarkers showed that removing amyloid improved CSF and plasma p-tau (p-tau181), downstream of amyloid in the AD pathology pathway. Tau PET analysis showed that lecanemab treatment slowed tau accumulation in the temporal lobe as well as improved total tau (t-tau) in compared to the placebo. As for biomarkers of neurodegeneration, lecanemab improved glial fibrillary acidic protein (GFAP) in plasma, a marker of astrocyte activation, and neurogranin in CSF, a marker of synaptic dysfunction, improved to normal levels by treatment, while there was no significant difference in neurofilament light chains in CSF or plasma between lecanemab and placebo.
Clarity AD Results in Context
AD is a progressive neurological disorder that severely impacts people living with the condition and their loved ones. With the increased global aging population, AD has become a critical issue for society and healthcare systems. New therapeutic agents that act on the disease pathology are needed. The treatment goals for early AD are to have sustained effects on cognitive function, activities of daily living and psychiatric symptoms, to maintain independence longer by slowing progression of the disease and to improve or maintain quality of life.
In Eisai’s confirmatory Clarity AD study, lecanemab demonstrated consistency of results across scales of cognition and function and subgroups (race, ethnicity, comorbidities). Lecanemab treatment showed 31% lower risk of converting to next stage of disease by Global CDR assessment (Hazard Ratio: 0.69). A slope analysis using CDR-SB based on observed data and extrapolation to 30 months showed that lecanemab takes 25.5 months to reach same level as placebo at 18 months, indicating a 7.5 month slowing of progression. Modeling simulations based on the phase 2 trial data suggest that lecanemab may slow the rate of disease progression by 2.5-3.1 years and has the potential to help people remain in the earlier stages of AD for a longer period of time. In addition, it was shown to maintain the health-related quality of life and reduce the burden on caregivers (23-56% reduction in score worsening). The convergence of evidence across cognition and function, disease progression, health related quality of life, and caregiver burden demonstrate that lecanemab treatment may provide meaningful benefits to patients, their care partners, physicians and society.
Eisai is hosting a live webcast of the scientific session featuring the lecanemab presentations, which can be viewed live on the investors section of the Eisai Co., Ltd. website. The content will be available on demand afterward.
Eisai serves as the lead of lecanemab development and regulatory submissions globally with both Eisai and Biogen co-commercializing and co-promoting the product and Eisai having final decision-making authority.
This release discusses investigational uses of an agent in development and is not intended to convey conclusions about efficacy or safety. There is no guarantee that such an investigational agent will successfully gain health authority approval.
1 CDR-SB is a numeric scale used to quantify the various severity of symptoms of dementia. Based on interviews of people living with AD and family/caregivers, qualified healthcare professionals assess cognitive and functional performance in six areas: memory, orientation, judgment and problem solving, community affairs, home and hobbies, and personal care. The total score of the six areas is the score of CDR-SB, and CDR-SB is also used as an appropriate item for evaluating the effectiveness of therapeutic drugs targeting the early stages of AD.
2 ADAS-Cog is the most common cognitive assessment instrument used in AD clinical trials all over the world. ADAS-Cog14 consists of 14 competencies: word recall, commands, constructional praxis, object and finger naming, ideational praxis, orientation, word recognition, remembering word recognition instructions, comprehension of spoken language, word finding difficulty, spoken language ability, delayed word recall, number cancellation, and maze task. ADAS-Cog has been used in clinical trials for earlier stages of AD including MCI.
3 Developed by Eisai, ADCOMS combines items from the ADAS-Cog scale for assessing cognitive functions, MMSE and the CDR scale for evaluating the severity of dementia to enable highly sensitive detection of changes in clinical functions of early AD symptoms and changes in memory
4 ADCS MCI-ADL assesses the competence of patients with MCI in activities of daily living (ADLs), based on 24 questions to the patient’s partner about actual recent activities of daily living.
Contacts
-
Eisai
MEDIA CONTACT:
Eisai Co., Ltd.
Public Relations Department
TEL: +81-(0)3-3817-5120
Eisai Inc. (U.S.)
Libby Holman
+ 1-201-753-1945
Libby_Holman@eisai.com
Eisai Europe, Ltd.
(Europe, Australia, New Zealand and Russia)
EMEA Communications Department
EMEA-comms@eisai.net
INVESTOR CONTACT:
Eisai Co., Ltd.
Investor Relations Department
TEL: +81-(0)70-8688-9685
-
Biogen
MEDIA CONTACT:
Biogen Inc.
Natacha Gassenbach
+ 1-857-777-6573
public.affairs@biogen.com
INVESTOR CONTACT:
Biogen Inc.
Mike Hencke
+ 1-781-464-2442
IR@biogen.com
[Notes to editors]
1. About Clarity AD
| Study title | A Study to Confirm Safety and Efficacy of Lecanemab in Participants With Early AD (Clarity AD) |
| Study population | 1,795 participants with mild cognitive impairment (MCI) due to AD and mild AD (collectively known as early AD) with confirmed presence of amyloid pathology in the brain in the global study, and an additional 111 subjects ongoing in China. |
| Treatment administered | 10 mg/kg bi-weekly of lecanemab |
| Duration of treatment | 18 months |
| Study locations | Japan, the U.S., Europe, China, South Korea, Canada, Australia, Singapore |
| Primary endpoint | Change from baseline in the Clinical Dementia Rating-Sum of Boxes (CDR-SB) at 18 months |
| Key secondary endpoints | Change From Baseline in Amyloid Positron Emission Tomography (PET) using Centiloids, AD Assessment Scale - Cognitive Subscale 14 (ADAS-Cog14*), AD Composite Score (ADCOMS**) and AD Cooperative Study-Activities of Daily Living Scale for Mild Cognitive Impairment (ADCS MCI-ADL***) at 18 months |
| Analysis Object | An efficacy analysis was conducted in a modified intention-to-treat population of 1,734 subjects (859 lecanemab group and 875 placebo group). The safety analysis was performed on all 1,795 randomized participants (lecanemab group: 898 placebo group: 897). |
- 2. About Lecanemab
Lecanemab is an investigational humanized monoclonal antibody for AD that is the result of a strategic research alliance between Eisai and BioArctic. Lecanemab selectively binds to neutralize and eliminate soluble, toxic amyloid-beta (Aβ) aggregates (protofibrils) that are thought to contribute to the neurodegenerative process in AD. As such, lecanemab may have the potential to have an effect on disease pathology and to slow down the progression of the disease. Currently, lecanemab is being developed as the only anti- Aβ antibody that can be used for the treatment of early AD without the need for titration. The Phase 2 clinical study (Study 201) in early AD subjects demonstrated reduction of brain Aβ accumulation (P<0.0001) and slowing of disease progression measured by ADCOMS (P<0.05) based on the pre-specified analysis at 18 months of treatment. The study did not achieve its primary outcome measure* at 12 months of treatment. The Study 201 open-label extension was initiated after completion of the Core period and a Gap period off treatment of 9-59 months (average of 24 months, n=180 from core study enrolled) to evaluate safety and efficacy and is underway.
Since July 2020 the Phase 3 clinical study (AHEAD 3-45) for individuals with preclinical AD, meaning they are clinically normal and have intermediate or elevated levels of amyloid in their brains, is ongoing. AHEAD 3-45 is conducted as a public-private partnership between the Alzheimer’s Clinical Trial Consortium that provides the infrastructure for academic clinical trials in AD and related dementias in the U.S, funded by the National Institute on Aging, part of the National Institutes of Health, Eisai and Biogen.
Since January 2022, the Tau NexGen clinical study for Dominantly Inherited AD (DIAD), that is conducted by Dominantly Inherited Alzheimer Network Trials Unit (DIAN-TU), led by Washington University School of Medicine in St. Louis, is ongoing.
Furthermore, Eisai has initiated a lecanemab subcutaneous dosing Phase 1 study.
In July 2022, the U.S. Food and Drug Administration (FDA) accepted Eisai’s Biologics License Application (BLA) for lecanemab under the accelerated approval pathway and granted Priority Review. The Prescription Drug User Fee Act action date (PDUFA) is set for January 6, 2023. The FDA has agreed that the results of Clarity AD can serve as the confirmatory study to verify the clinical benefit of lecanemab. In an effort to secure traditional FDA approval for lecanemab as soon as possible, Eisai submitted the BLA through the FDA’s Accelerated Approval Pathway so that the agency could complete its review of all lecanemab data with the exception of the data from the confirmatory Clarity AD study. In March 2022, Eisai began submitting application data, with the exception of Clarity AD data, to Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) under the prior assessment consultation system. Eisai will discuss the results of Clarity AD study with regulatory authorities in the U.S., Japan and Europe with the aim to file for traditional approval in the U.S. and for marketing authorization applications in Japan and Europe by the end of Eisai’s FY2022, which ends March 31, 2023.
* An 80% or higher estimated probability of demonstrating 25% or greater slowing in clinical decline at 12 months treatment measured by ADCOMS from baseline compared to placebo.
- 3. About the Collaboration between Eisai and Biogen for AD
Eisai and Biogen have been collaborating on the joint development and commercialization of AD treatments since 2014. Eisai serves as the lead of lecanemab development and regulatory submissions globally with both companies co-commercializing and co-promoting the product and Eisai having final decision-making authority.
- 4. About the Collaboration between Eisai and BioArctic for AD
Since 2005, Eisai and BioArctic have had a long-term collaboration regarding the development and commercialization of AD treatments. Eisai obtained the global rights to study, develop, manufacture and market lecanemab for the treatment of AD pursuant to an agreement with BioArctic in December 2007. The development and commercialization agreement on the antibody lecanemab back-up was signed in May 2015.
- 5. About Eisai Co., Ltd.
Eisai’s Corporate Concept is “to give first thought to patients and people in the daily living domain, and to increase the benefits that health care provides.” Under this Concept (also known as human health care (hhc) Concept), we aim to effectively achieve social good in the form of relieving anxiety over health and reducing health disparities. With a global network of R&D facilities, manufacturing sites and marketing subsidiaries, we strive to create and deliver innovative products to target diseases with high unmet medical needs, with a particular focus in our strategic areas of Neurology and Oncology.
In addition, we demonstrate our commitment to the elimination of neglected tropical diseases (NTDs), which is a target (3.3) of the United Nations Sustainable Development Goals (SDGs), with working on various activities together with global partners.
For more information about Eisai, please visit www.eisai.com (for global headquarters: Eisai Co., Ltd.), and connect with us on Twitter @Eisai_SDGs.
- 6. About Biogen
As pioneers in neuroscience, Biogen discovers, develops, and delivers worldwide innovative therapies for people living with serious neurological diseases as well as related therapeutic adjacencies. One of the world’s first global biotechnology companies, Biogen was founded in 1978 by Charles Weissmann, Heinz Schaller, Sir Kenneth Murray, and Nobel Prize winners Walter Gilbert and Phillip Sharp. Today, Biogen has a leading portfolio of medicines to treat multiple sclerosis, has introduced the first approved treatment for spinal muscular atrophy, and developed the first and only approved treatment to address a defining pathology of Alzheimer’s disease. Biogen is also commercializing biosimilars and focusing on advancing one of the industry’s most diversified pipelines in neuroscience that will transform the standard of care for patients in several areas of high unmet need.
We routinely post information that may be important to investors on our website at www.biogen.com. Follow us on social media - Twitter, LinkedIn, Facebook, YouTube.
Biogen Safe Harbor
This news release contains forward-looking statements, including statements made pursuant to the safe harbor provisions of the Private Securities Litigation Reform Act of 1995, about the potential clinical effects of lecanemab; the potential benefits, safety and efficacy of lecanemab; potential regulatory discussions, submissions and approvals and the timing thereof; the treatment of Alzheimer’s disease; the anticipated benefits and potential of Biogen’s collaboration arrangements with Eisai; the potential of Biogen’s commercial business and pipeline programs, including lecanemab; and risks and uncertainties associated with drug development and commercialization. These statements may be identified by words such as “aim,” “anticipate,” “believe,” “could,” “estimate,” “expect,” “forecast,” “intend,” “may,” “plan,” “possible,” “potential,” “will,” “would” and other words and terms of similar meaning. Drug development and commercialization involve a high degree of risk, and only a small number of research and development programs result in commercialization of a product. Results in early-stage clinical studies may not be indicative of full results or results from later stage or larger scale clinical studies and do not ensure regulatory approval. You should not place undue reliance on these statements or the scientific data presented.
These statements involve risks and uncertainties that could cause actual results to differ materially from those reflected in such statements, including without limitation unexpected concerns that may arise from additional data, analysis or results obtained during clinical studies, including the Clarity AD clinical trial and AHEAD 3-45 study; the occurrence of adverse safety events; risks of unexpected costs or delays; the risk of other unexpected hurdles; regulatory submissions may take longer or be more difficult to complete than expected; regulatory authorities may require additional information or further studies, or may fail or refuse to approve or may delay approval of Biogen’s drug candidates, including lecanemab; actual timing and content of submissions to and decisions made by the regulatory authorities regarding lecanemab; uncertainty of success in the development and potential commercialization of lecanemab; failure to protect and enforce Biogen’s data, intellectual property and other proprietary rights and uncertainties relating to intellectual property claims and challenges; product liability claims; third party collaboration risks; and the direct and indirect impacts of the ongoing COVID-19 pandemic on Biogen’s business, results of operations and financial condition. The foregoing sets forth many, but not all, of the factors that could cause actual results to differ from Biogen’s expectations in any forward-looking statement. Investors should consider this cautionary statement as well as the risk factors identified in Biogen’s most recent annual or quarterly report and in other reports Biogen has filed with the U.S. Securities and Exchange Commission. These statements are based on Biogen’s current beliefs and expectations and speak only as of the date of this news release. Biogen does not undertake any obligation to publicly update any forward-looking statements, whether as a result of new information, future developments or otherwise.
Our news releases are intended to disclose corporate information and are not intended for the promotion or advertising of prescription pharmaceuticals or investigational products, nor are they intended to provide medical advice.
Please note that the information contained in news releases is current as of the date of publication.