- For Print
- June 25, 2013
Eisai Co., Ltd. (Headquarters: Tokyo, President & CEO: Haruo Naito, “Eisai”) announced today that it has decided to suspend temporarily commercial distribution of the antiepileptic drug, AMPA receptor antagonist Fycompa® (perampanel) in Germany based on its belief that the German Federal Joint Committee (G-BA) failed to appropriately assess the value of Fycompa as an innovative new treatment in an additional benefit assessment conducted after German marketing approval was granted for the drug last year. Eisai's primary concern in the country will continue to be for patients requiring Fycompa treatment and the company will provide a Patient Access Program to ensure that patients are still able to access the drug after the temporary suspension comes into effect.
According to the additional benefit assessment, which was carried out based on Germany's Arzneimittelmarkt-Neuordnungsgesetz (Act on the Reform of the Market for Medical Products, or “AMNOG”), the G-BA concluded in March 2013 that the submitted assessment data comparing perampanel to two other treatments as defined by the G-BA itself were insufficient and that the additional benefit of perampanel was thus unproven. The German Society for Epileptology and the German Society for Neurology have both commented on the methodology used in the comparison, pointing out that assessments of adjunctive treatments for patients with refractory epilepsy in which monotherapy treatment was inadequate should consider whether the adjunctive treatment offers an additional benefit as an add-on therapy in patients who are resistant to standard treatments. Both societies also agree that assessments comparing such novel drugs to existing therapies chosen by the G-BA are inappropriate in that they do not reflect the clinical situation of refractory epilepsy. Furthermore, the two organizations reaffirm that for patients with refractory epilepsy, there is a pressing need for new drugs that can offer improved efficacy in terms of seizure management and reducing side effects, and that the current clinical situation for this type of epilepsy in particular requires an innovative treatment such as Fycompa, which possesses a novel mechanism of action.
Meanwhile, the additional benefit assessment system has also undergone partial revision in Germany, with a recent amendment to the German Law on Medicines addressing the issues outlined above. Eisai will be closely watching for further related developments in the country and is positive about being able to resolve the situation in regard to its temporary sales suspension of Fycompa in Germany, and plans to submit perampanel as soon as possible for a reassessment based on appropriate assessment methodology that recognizes the drug's efficacy and innovation.
Fycompa was approved by the European Commission in July 2012 as an adjunctive treatment for partial-onset seizures with or without secondarily generalized seizures in patients with epilepsy aged 12 years and older. It was launched in Germany in September of the same year and has been used by 3,000 patients in that country to date. The drug is currently available in seven European countries. In the United States, Fycompa was approved by the U.S. Food and Drug Administration (FDA) in October 2012 and is planned for launch once the U.S. Drug Enforcement Administration (DEA) has completed its scheduled investigation under the Controlled Substances Act.
Eisai remains committed to seeking out an accurate understanding and assessment from the relevant authorities regarding the value of Fycompa in order to continue to ensure delivery of the drug to patients with partial-onset seizures in Germany in need of innovative new therapies.
[ Please refer to the following notes for further information on the treatment of epilepsy,
a glossary of terms, and outlines on Fycompa and related perampanel Phase III studies. ]
Media Inquiries:
Public Relations Department,
Eisai Co., Ltd.
+81-(0)3-3817-5120
< Notes to editors >
1. About the Treatment of Epilepsy
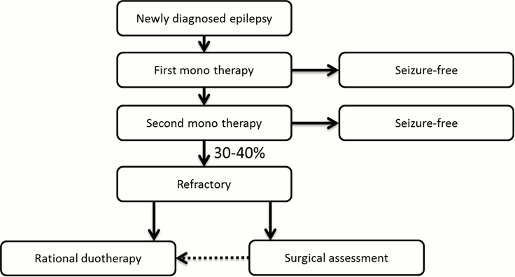
The goal of treating patients with epilepsy is seizures-free. Once epilepsy is diagnosed, a patient usually begins monotherapy (single-agent) treatment with an antiepileptic drug. In cases where the treatment's efficacy on seizure control is inadequate, the initial treatment can typically be replaced with a different monotherapy. In approximately 30-40% of patients, however, seizure control is not achieved even after undergoing two different monotherapy treatments and in these circumstances the patient can then opt to switch to a treatment that includes an adjunctive therapy. In these cases, it is drugs like Fycompa® that are approved as adjunctive therapies in treating epilepsy that are used as “add-ons” to the patient's existing monotherapy treatment. Furthermore, depending on the patient's symptoms, there may be multiple “add-on” adjunctive therapies being used at one time.
2. Glossary of Terms
- 1)German Federal Joint Committee (G-BA)
The German Federal Joint Committee (Gemeinsamer Bundesausschuss, G-BA) is the highest decision-making body of the joint self-government of physicians, dentists, hospitals and health insurance funds in Germany. It issues directives for the benefit catalog of statutory health insurance funds (GKV) and thus specifies which drugs and medical services are reimbursed by the GKV.
- 2)About Germany's Act on the Reform of the Market for Medical Products (AMNOG)
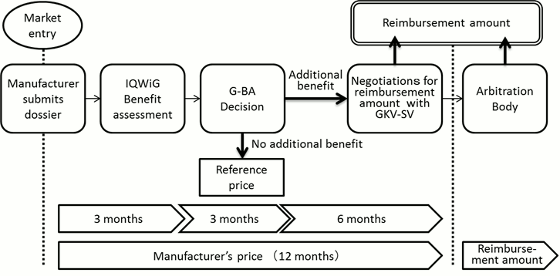
In Germany, the enactment of the Act on the Reform of the Market for Medical Products (Arzneimittelmarkt-Neuordnungsgesetz, AMNOG) came into effect on January 2011. Under this amendment, all eligible new drugs launched on the German market must undergo an additional benefit assessment conducted by the G-BA, with later price negotiations to be based on this assessment and a reimbursement price to be decided within one year from the drug's launch.
Furthermore, when a new drug is launched, the pharmaceutical company must submit to the G-BA a benefit dossier demonstrating the drug's additional benefit over a comparator as defined by the G-BA itself. The G-BA then usually commissions the country's Institute for Quality and Efficiency in Health Care (IQWiG) to evaluate the dossier to decide whether any additional benefit exists over the comparator and the results are subsequently published. The pharmaceutical company is next given an opportunity to comment at a hearing on the IQWiG's evaluation, after which the G-BA carries out its final decision regarding any additional benefit of the drug.
If an additional benefit is recognized by the G-BA, the drug proceeds to the price negotiation stage with the lead association of the German sick funds (GKV-SV), and a reimbursement price is decided based on the level of additional benefit as decided by the G-BA. On the other hand, a drug deemed to offer no recognized additional benefit is designated a reference price group as well as a reimbursement price based on the price of the comparator used during the benefit assessment.
3. About Fycompa® (Perampanel)
Fycompa (perampanel), a novel chemical entity discovered and undergoing development by Eisai, is a highly selective, non-competitive AMPA -type glutamate receptor antagonist. Perampanel is the first antiepileptic treatment to reduce neuronal hyperexcitation associated with seizures by targeting glutamate activity at post-synaptic AMPA receptors. Perampanel has demonstrated broad-spectrum antiseizure effects in Phase II and III studies. The agent has received a positive opinion from the CHMP for the adjunctive treatment of partial-onset seizures in Europe, is currently under regulatory review for NDA in the United States, and is also being evaluated in Phase III studies in Japan. Furthermore, Eisai is conducting a global Phase III studies for generalized epilepsy and plans to conduct further studies for usage as monotherapy in the treatment of partial-onset seizures, Lennox-Gastaut syndrome and other forms of epilepsy as it seeks to expand the range of indications for which the drug is approved.
4. About Perampanel Phase III Studies
The clinical development plan for perampanel consisted of three global Phase III studies (Studies 306, 305 and 304) in which a total of 1,480 epilepsy patients with partial-onset seizures ages 12 years and older participated. The key goal of Study 306 was to identify the minimal effective dose and included four treatment arms (placebo, 2 mg, 4 mg, and 8 mg). Studies 304 and 305 included three arms (placebo, 8 mg, and 12 mg) and were to evaluate a more extended dose range.
The studies were similar in design: global, randomized, double-blind, placebo-controlled, dose-escalation, parallel-group studies. The primary and secondary endpoints were the same in all the studies: percentage change in seizure frequency, 50% responder rate, percentage reduction of complex partial plus secondarily generalized seizures, and evaluation for dose response. The primary endpoint for the EMA is 50% responder rate and the FDA is median percent change in seizure frequency.
Results of the studies are as follows.
- 1)Study 306
- The 50% responder rates compared to placebo for the ITT (intention-to-treat) population were:
2 mg = 20.6% (p = 0.4863), 4 mg = 28.5% (p = 0.0132), 8 mg = 34.9% (p = 0.0003) versus 17.9% with placebo. - The median percent change in seizure frequency for the ITT population were:
2 mg = -13.6% (p = 0.4197), 4 mg = -23.3% (p = 0.0026), 8 mg = -30.8% (p < 0.0001) versus -10.7% with placebo - The most frequent treatment-emergent adverse events were dizziness, headache and somnolence.
- The 50% responder rates compared to placebo for the ITT (intention-to-treat) population were:
- 2)Study 305
- The 50% responder rates compared to placebo for the ITT population were:
8 mg = 33.3% (p = 0.0018), 12 mg = 33.9% (p = 0.0006) versus 14.7% with placebo. - The median percent change in seizure frequency for the ITT population were:
8 mg = -30.5% (p = 0.0008), 12 mg = -17.6% (p = 0.0105) versus -9.7% with placebo - The most reported adverse events were dizziness, fatigue, headache and somnolence.
- The 50% responder rates compared to placebo for the ITT population were:
- 3)Study 304
- The 50% responder rates compared to placebo for the ITT population were:
8 mg = 37.6% (p = 0.0760), 12 mg = 36.1% (p = 0.0914) versus 26.4% with placebo - The median percent change in seizure frequency for the ITT population were:
8 mg = -26.3% (p = 0.0261), 12 mg = -34.5% (p = 0.0158) and placebo = -21.0% - The most common side effects were dizziness, somnolence, irritability, headache, falls and ataxia.
- The 50% responder rates compared to placebo for the ITT population were: